

Abstract
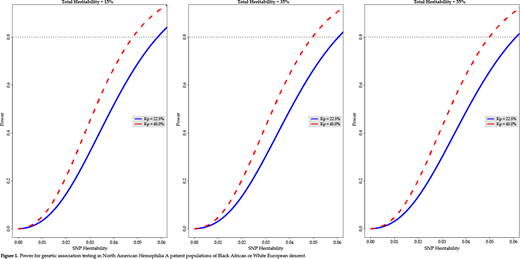
Here we apply state-of-the-art statistical genetic approaches toward investigating the genetic architecture of factor VIII (FVIII) inhibitor (FEI) development in Hemophilia A (HA). A total of 442 North American HA patients (237 Whites and 205 Blacks; 88% severely affected) enrolled in the PATH Study were: 1) ImmunoChip genotyped at ~167,000 single nucleotide polymorphisms (SNPs) in genes previously implicated in autoimmune disease risk; 2) Evaluated by DNA sequencing and assays for the recurrent intron (I)1 and I22 inversions to identify their causative F8 mutations; and 3) Tested with the Bethesda assay to determine their FEI status. The ImmunoChip genotypes were used to construct a genetic relationship matrix (GRM), denoted by K, following our previously published method,1 and the F8 sequence data along with results from the I1 and I22 inversion assays were used to construct a shared F8-mutation matrix, denoted by F. We analyzed a dichotomous FEI variable under the statistical genetic threshold/liability model (a probit regression in the fixed effects) in conjunction with a variance components model for the FEI liability phenotypic covariance matrix, denoted by P, to model potentially important random effects. For the latter, we specifically assumed independent additive genetic, F8-mutation, and residual environmental random effects. By the independence assumption, the covariance matrix is then decomposable as a sum of the additive genetic (Va), F8-mutation (Vf), and residual environmental (Ve) variances respectively structured by K, F, and the identity matrix I. The variance component model is given as: P = K*Va + F*Vf + I*Ve. Heritability, denoted by h2, is defined as the ratio of Va to the total phenotypic variance (Vp): h2 = Va / Vp. We can further speak of the total heritability given as: h2t = h2r + h2f + h2snp, where the subscripts t, r, f, and snp respectively denote total, residual additive genetic, F8-mutation-specific, and SNP heritabilities. Using eigenstructure methods,2 we can compute power under a simpler model in which Va and Vf are combined as a single variance component. We computed power to detect genetic association as measured by SNP-specific heritability for a set of 403 SNPs in or near 14 candidate immune response genes previously implicated in FEI risk. To account for multiple hypothesis testing, power was computed at the Bonferroni-adjusted significance level of 0.05/403 = 1.2 × 10-4. Under the simplified model, we computed the statistical power to detect causal SNPs for our sample and study design for the sample FEI prevalences, denoted by Kp, for Whites (22.5%) and Blacks (45%), across a range of total heritabilities, h2t = 15%, 35%, and 55%, where the lattermost total heritability was observed for FEI liability in the current study (Figure 1). It should be noted that because the liability heritability is known to be biased upward, we applied the Dempster-Lerner correction to both the total and SNP-specific heritabilities.3 Close inspection of Figure 1 reveals that varying h2t from 15% to 35% to 55% results in slight decreases in power due to the decreasing ratio of the SNP-specific heritability to the total heritability. However, as seen in all three panels, the more important determinant of power is clearly the FEI prevalence in that the power curve for a Kp of 45% is associated with greater power than the power curve for a Kp of 22.5% across the range of total heritabilities examined. As seen in Figure 1, we have adequate power to detect SNP heritabilities as low as 5% and 6%, respectively, for a Kp of 45% and 22.5%. As noted above, we observed a FEI liability total heritability of 55% consisting of a 47% residual additive genetic heritability (p = 0.019) and 8% F8-mutation specific heritability (p = 0.005). This is the first study to use a GRM based on genotype data and a shared causal F8 mutation matrix to model additive genetic and F8-mutation specific effects.
Almeida M, Peralta J, Farook V, …, Blangero J. Pedigree-based random effect tests to screen gene pathways. BMC Proc. 2014; 8(Suppl 1 Genetic Analysis Workshop): S100.
Blangero J, Diego VP, Dyer T, …, Göring H. A kernel of truth: statistical advances in polygenic variance component models for complex human pedigrees. Adv Genetics. 2013; 81: 1-31.
Glahn D, Williams J, McKay D, …, Blangero J. Discovering schizophrenia endophenotypes in randomly ascertained pedigrees. Biol Psychiatry. 2015; 77(1): 75-83.
Chitlur:Baxter, Bayer, Biogen Idec, and Pfizer: Honoraria; Novo Nordisk Inc: Consultancy. Dinh:Haplomics Biotechnology Corporation: Employment, Equity Ownership. Howard:Haplomics Biotechnology Corporation: Equity Ownership, Other: Chief Scientific Officer, Patents & Royalties: Patent applications and provisional patent applications ; CSL Behring: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal